如何去學一個R包(下)
- 2020 年 3 月 30 日
- 筆記
回顧
檢查偽時間基因表達的變化
FateID還提供偽時間基因表達變化的可視化和分析功能。為此,可以提取具有朝向目標簇的命運偏差的細胞。principal curve 分析以偽時間順序返回沿著分化軌跡的所有細胞。例如,可以通過以下命令提取以偽時間順序向簇6具有命運偏差的單元:
n <- pr$trc[["t6"]]
或者用其他的偽時間排序的方法,比如Monocle2 (Qiu等人2017)或擴散偽時間6。這個包可以通過擴散偽時間按cell命運概率和後續偽時間排序執行過濾:
trc <- dptTraj(x,y,fb,trthr=.25,distance="euclidean",sigma=1000) #n <- trc[["t6"]]
如果trthr沒有給出最小的細胞命運概率的閾值,則僅使用具有朝向特定目標聚類的具有顯著命運偏差的細胞來填充相應的軌跡。該函數以偽時間順序返回包含推斷的差異軌跡上的所有單元的每個目標簇的向量列表。
在第二步中,定義輸入表達數據:
v <- intestine$v fs <- filterset(v,n=n,minexpr=2,minnumber=1)
在這裡,我們使用未經挑選的全部表達數據v。第二個參數是以偽時間順序對應於表達表的有效行名的細胞id的向量。
該filterset功能可用於從隨後的分析中消除軌跡上的低表達基因,並且具有兩個額外的參數來丟棄基因,所述基因表達量應高於minexpr以及至少在minnumber個細胞中表達。該函數以與輸入向量n中相同的順序返回過濾的表達式數據集,其中基因作為行,單元格作為列。在第三步,計算偽時間表達譜的自組織映射(SOM):
s1d <- getsom(fs,nb=1000,alpha=.5)
此映射提供了一組類似的表達譜到模組中。第一個輸入參數也是表達數據。在這種情況下,我們使用filterset函數生成的過濾表達表來保留僅在所考慮的軌跡上表達的基因。在通過平滑參數alpha的局部回歸進行平滑化之後,計算沿著感興趣的分化軌跡的偽時間表達譜。
此函數返回以下三個列表:som包中的som功能返回som對象,具有平滑和標準化的表達數據x,以及z分數轉換的偽時間表達譜轉換的z-score的數據集zs。
然後,SOM由另一個函數處理,以將SOM的節點分組為更大的模組,並生成用於展示的 z-score變換和分箱表達式數據集:
ps <- procsom(s1d,corthr=.85,minsom=3)
The output of the processed SOM can be plotted using the plotheatmap function. First, in order to highlight the clustering partition y, the same color scheme as used in plotFateMap can be generated (alternatively, both functions can be executed with an arbitrary user-defined scheme):
第一個輸入參數是通過函數getsom計算的SOM給出。該函數有兩個額外的輸入參數來控制SOM節點分組為更大的模組。參數corthr定義了相關閾值。如果SOM中相鄰節點的偽時間表達譜平均歸一化的z-score的相關性超過該閾值,則相鄰節點的基因被合併到更大的模組中。僅保留至少具有minsom基因的模組。該函數返回各種數據集的列表,其中包括規範化,z-score轉換或將基因分配到SOM模組的分箱表達式(有關詳細資訊,請參閱幫助頁面)。
可以使用plotheatmap函數繪製已處理SOM的輸出。首先,為了突出顯示聚類分區y,plotFateMap可以生成與使用的顏色方案相同的顏色方案(或者,可以使用用戶定義的任意方案執行這兩個功能):
set.seed(111111) fcol <- sample(rainbow(max(y)))
現在,procsom的不同輸出數據可以繪製。
從SOM派生的所有模組的平均z分數:
plotheatmap(ps$nodes.z, xpart=y[n], xcol=fcol, ypart=unique(ps$nodes), xgrid=FALSE, ygrid=TRUE, xlab=FALSE)

SOM模組中每個基因的z-score概況:
plotheatmap(ps$all.z, xpart=y[n], xcol=fcol, ypart=ps$nodes, xgrid=FALSE, ygrid=TRUE, xlab=FALSE)

SOM模組排序的每個基因的標準化表達譜:
plotheatmap(ps$all.e, xpart=y[n], xcol=fcol, ypart=ps$nodes, xgrid=FALSE, ygrid=TRUE, xlab=FALSE)

每個基因的二值化表達譜(z-score <-1,-1<z-score <1,z-score="" style="font-size: inherit; color: inherit; line-height: inherit;">1):
plotheatmap(ps$all.b, xpart=y[n], xcol=fcol, ypart=ps$nodes, xgrid=FALSE, ygrid=TRUE, xlab=FALSE)
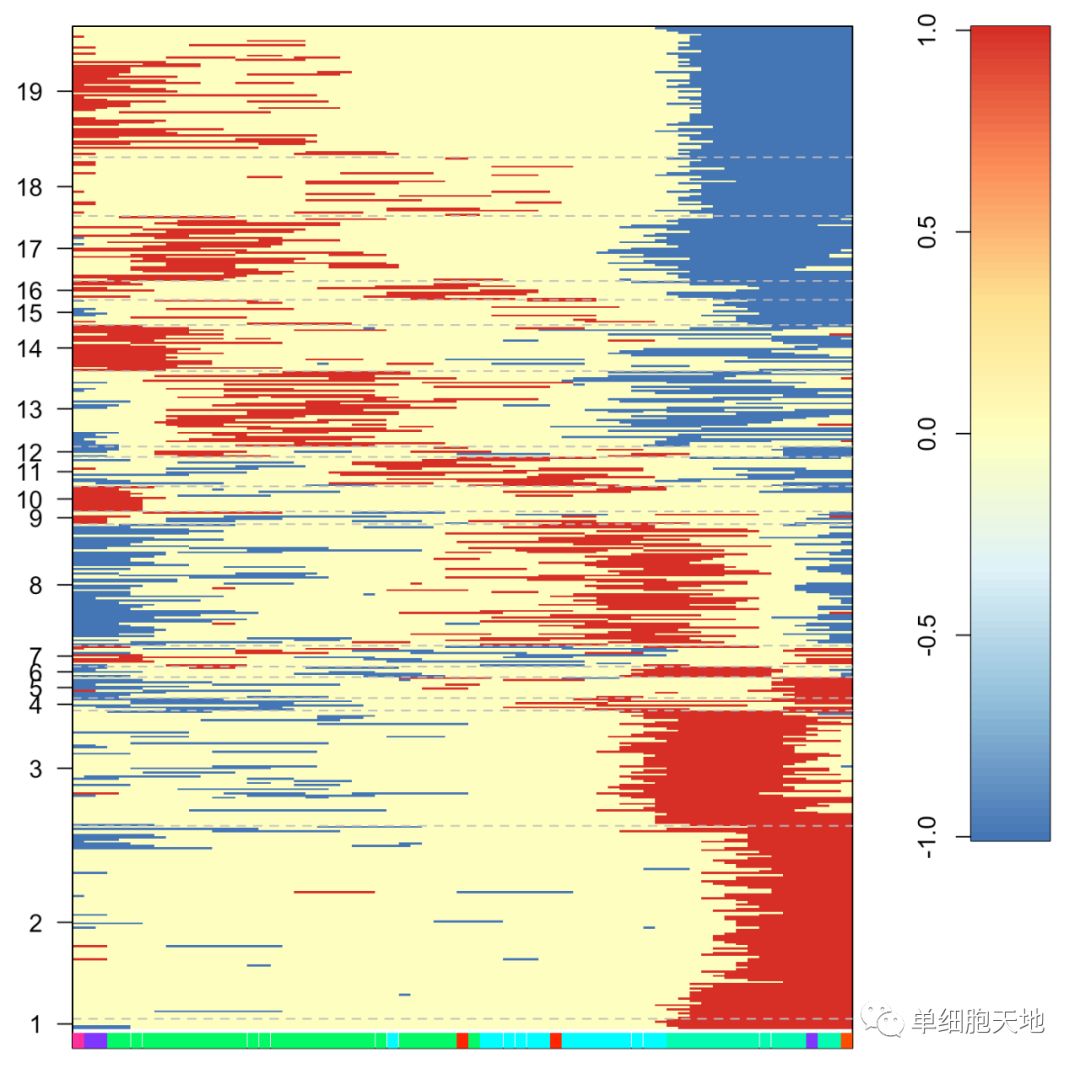
為了檢查SOM的各個模組內的基因,可以在給定模組的數量的情況下提取這些基因。模組編號包含在procsom函數的返回參數節點中,並且可以提取,例如,對於模組編號1:
g <- names(ps$nodes)[ps$nodes == 1]
該組的平均偽時間表達譜可以通過函數plotexpression繪製:
plotexpression(fs, y, g, n, col=fcol, name="Node 1", cluster=FALSE, alpha=.5, types=NULL)

以同樣的方式可以繪製單個基因的表達譜,例如:
plotexpression(fs, y, "Clca4__chr3", n, col=fcol, cluster=FALSE, alpha=.5, types=NULL)

也可以使用types參數將數據點突出顯示為特定符號,例如反映批次:
plotexpression(fs, y, g, n, col=fcol, name="Node 1", cluster=FALSE, types=sub("\_\d+","",n))

差異基因表達分析
FateID還提供了差異基因表達分析的功能。該功能實現了類似於DESeq (Anders和Huber 2010)的方法,通過該方法將負二項分布擬合到兩個群體中的轉錄物計數,例如表示兩種不同的細胞類型。默認情況下,該函數應在標準化數據上執行,並根據對數轉換的轉錄計數方差的多項式擬合估計色散參數,作為對數轉換平均表達式的函數。這是在輸入數據上完成的,要麼在彙集來自兩個群體的細胞之後,要麼分別在每個群體上彙集細胞。或者,還可以提供方差 – 平均依賴性的函數作為輸入參數,例如在RaceID2中計算的依賴性(Grun et al.2016)異常值識別。作為另一種選擇,該功能還可以執行標準DESeq2 (Love,Huber和Anders 2014)差異表達分析。
為了運行分析,表達數據與cell IDs向量對應於此數據集的列名子集要一起輸入。
例如,該功能可用於鑒定原始簇3,4,5(祖細胞區室)中細胞之間差異表達的基因,這些細胞偏向於腸細胞(簇6)或杯狀細胞(簇13),因為命運概率> 0.5:
thr <- .5 a <- "t13" b <- "t6" cl <- c(3,4,5) A <- rownames(fb$probs)[fb$probs[,a] > thr] A <- A[y[A] %in% cl] B <- rownames(fb$probs)[fb$probs[,b] > thr] B <- B[y[B] %in% cl] de <- diffexpnb(v,A=A,B=B,DESeq=FALSE,norm=FALSE,vfit=NULL,locreg=FALSE)
在這裡,我們使用沒有進行特徵選擇的完整表達數據作為輸入。不執行標準化,因為v已經按大小標準化。利用所選擇的輸入參數,diffexpnb將基於RaceID2(Grun等人2016)中導出的方差 – 均值依賴性推斷兩個群體的負二項轉錄物水平分布的分散參數。或者,如果locreg等於TRUE,則可以使用局部回歸。另一種選擇是運行DESeq2 (Love,Huber和Anders 2014)分析,如果DESeq是TRUE的話。該函數返回包含三個部分的列表。差異基因表達分析的結果存儲在res組件中,res組件是顯示兩組平均表達的數據框,兩組之間的倍數變化和log2倍數變化,差異表達式的p值(pval)和Benjamini-Hochberg糾正了錯誤發現率(padj)。
結果可以通過以下函數繪製:
plotdiffgenesnb(de,mthr=-4,lthr=0,Aname=a,Bname=b,padj=FALSE)

檢查基因表達的命運偏見
為了能夠進一步檢查命運特異性標記基因表達,FateID提供了在散點圖中繪製兩個基因彼此表達水平的功能。例如,這些基因可以代表兩種不同譜系的標記,並且該功能揭示這些基因的表達域是否落入不同的簇中,或者它們是否與命運偏向特定譜系相關。為了可視化基因表達域和細胞簇起源的相關性,gene2gene可以用基因表達數據框和聚類分區作為輸入來調用該函數:
gene2gene(intestine$v,intestine$y,"Muc2__chr7","Apoa1__chr9")
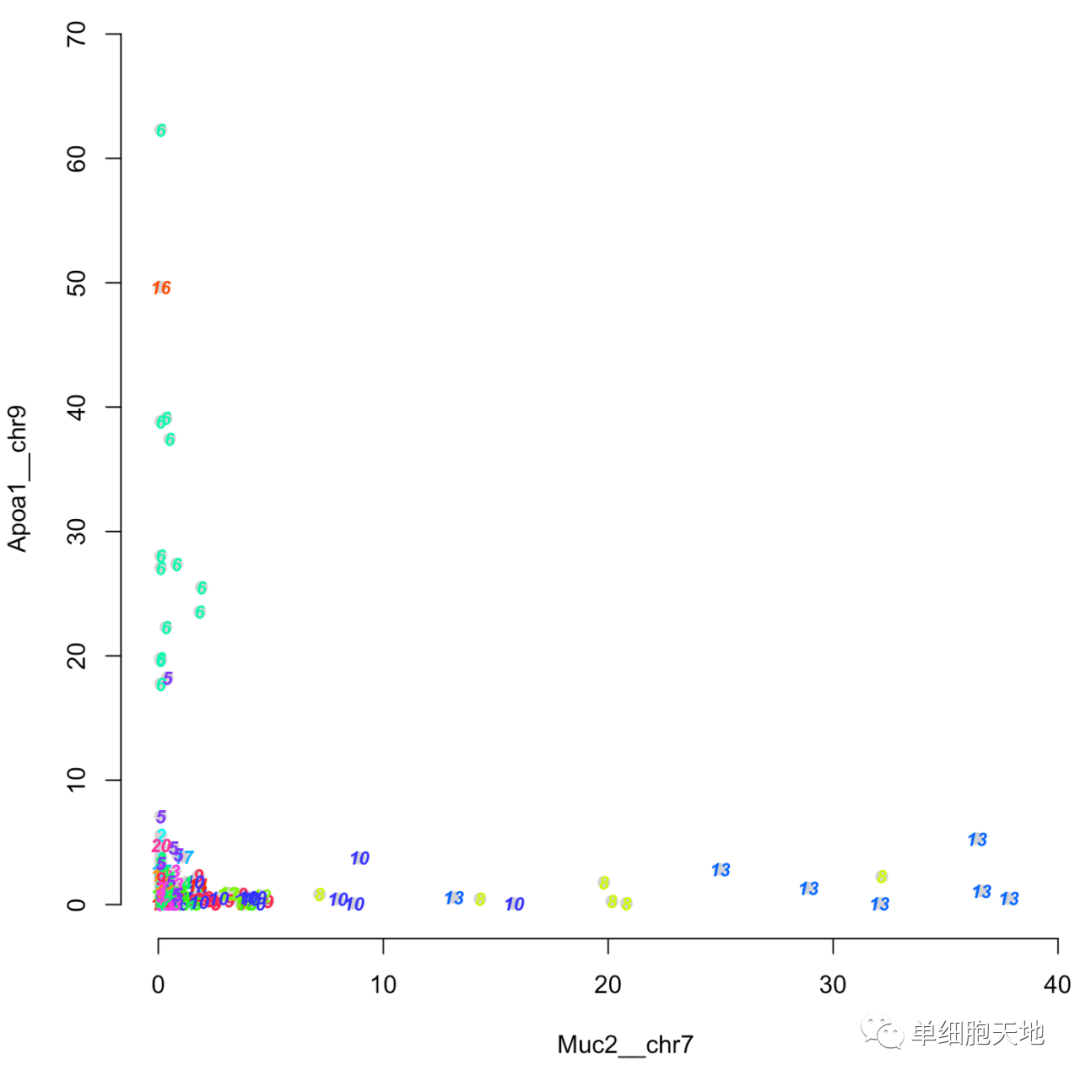
如果向特定的目標群的命運偏移,例如群集6,fateBias的輸出fb和fb$probs的列名對應於目標群集,即t6,需要作為附加輸入參數:
gene2gene(intestine$v, intestine$y, "Muc2__chr7", "Apoa1__chr9", fb=fb, tn="t6", plotnum=FALSE)

為了識別在分化的所有階段參與早期啟動和命運決定的基因,提取具有高重要性值的所有基因(參見randomForest包中的randomForest函數的詳細資訊)以分類到至少在一次隨機森林迭代中給定的目標聚類中:
k <- impGenes(fb,"t6")

此函數需要函數fateBias的輸出對象和目標集群的數量,並與t連接作為輸入。其他可選參數指定重要性值的截止值(參見手冊頁)。它返回含有兩個對象的列表,具有所有基因的平均重要性值的一個數據集,這裡所述基因至少在迭代中作為行或作為列通過閾值,以及具有重要性值的標準偏差的相應數據集。
該函數還繪製了重要值的熱圖,其中基因進行了層次聚類。
References
Anders, S., and W. Huber. 2010. 「Differential expression analysis for sequence count data.」 Genome Biol. 11 (10): R106.
Angerer, P., L. Haghverdi, M. Buttner, F. J. Theis, C. Marr, and F. Buettner. 2016. 「destiny: diffusion maps for large-scale single-cell data in R.」 Bioinformatics 32 (8): 1241–3.
Breiman, L. 2001. 「Random Forests.」 Mach. Learn. 45 (1): 5–32.
Grun, D., M. J. Muraro, J. C. Boisset, K. Wiebrands, A. Lyubimova, G. Dharmadhikari, M. van den Born, et al. 2016. 「De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data.」 Cell Stem Cell 19 (2): 266–77.
Herman, J. S., Sagar, and D. Grün. 2018. 「FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data.」 Nat. Methods 15 (5): 379–86.
Love, M. I., W. Huber, and S. Anders. 2014. 「Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.」 Genome Biol.15 (12): 550.
Maaten, L. van der, and G. Hinton. 2008. 「Visualizing Data using t-SNE.」 J. Mach. Learn. 9: 2570–2605.
Qiu, X., A. Hill, J. Packer, D. Lin, Y. A. Ma, and C. Trapnell. 2017. 「Single-cell mRNA quantification and differential analysis with Census.」 Nat. Methods 14 (3): 309–15.


