如何去学一个R包(下)
- 2020 年 3 月 30 日
- 笔记
回顾
检查伪时间基因表达的变化
FateID还提供伪时间基因表达变化的可视化和分析功能。为此,可以提取具有朝向目标簇的命运偏差的细胞。principal curve 分析以伪时间顺序返回沿着分化轨迹的所有细胞。例如,可以通过以下命令提取以伪时间顺序向簇6具有命运偏差的单元:
n <- pr$trc[["t6"]]
或者用其他的伪时间排序的方法,比如Monocle2 (Qiu等人2017)或扩散伪时间6。这个包可以通过扩散伪时间按cell命运概率和后续伪时间排序执行过滤:
trc <- dptTraj(x,y,fb,trthr=.25,distance="euclidean",sigma=1000) #n <- trc[["t6"]]
如果trthr没有给出最小的细胞命运概率的阈值,则仅使用具有朝向特定目标聚类的具有显著命运偏差的细胞来填充相应的轨迹。该函数以伪时间顺序返回包含推断的差异轨迹上的所有单元的每个目标簇的向量列表。
在第二步中,定义输入表达数据:
v <- intestine$v fs <- filterset(v,n=n,minexpr=2,minnumber=1)
在这里,我们使用未经挑选的全部表达数据v。第二个参数是以伪时间顺序对应于表达表的有效行名的细胞id的向量。
该filterset功能可用于从随后的分析中消除轨迹上的低表达基因,并且具有两个额外的参数来丢弃基因,所述基因表达量应高于minexpr以及至少在minnumber个细胞中表达。该函数以与输入向量n中相同的顺序返回过滤的表达式数据集,其中基因作为行,单元格作为列。在第三步,计算伪时间表达谱的自组织映射(SOM):
s1d <- getsom(fs,nb=1000,alpha=.5)
此映射提供了一组类似的表达谱到模块中。第一个输入参数也是表达数据。在这种情况下,我们使用filterset函数生成的过滤表达表来保留仅在所考虑的轨迹上表达的基因。在通过平滑参数alpha的局部回归进行平滑化之后,计算沿着感兴趣的分化轨迹的伪时间表达谱。
此函数返回以下三个列表:som包中的som功能返回som对象,具有平滑和标准化的表达数据x,以及z分数转换的伪时间表达谱转换的z-score的数据集zs。
然后,SOM由另一个函数处理,以将SOM的节点分组为更大的模块,并生成用于展示的 z-score变换和分箱表达式数据集:
ps <- procsom(s1d,corthr=.85,minsom=3)
The output of the processed SOM can be plotted using the plotheatmap function. First, in order to highlight the clustering partition y, the same color scheme as used in plotFateMap can be generated (alternatively, both functions can be executed with an arbitrary user-defined scheme):
第一个输入参数是通过函数getsom计算的SOM给出。该函数有两个额外的输入参数来控制SOM节点分组为更大的模块。参数corthr定义了相关阈值。如果SOM中相邻节点的伪时间表达谱平均归一化的z-score的相关性超过该阈值,则相邻节点的基因被合并到更大的模块中。仅保留至少具有minsom基因的模块。该函数返回各种数据集的列表,其中包括规范化,z-score转换或将基因分配到SOM模块的分箱表达式(有关详细信息,请参阅帮助页面)。
可以使用plotheatmap函数绘制已处理SOM的输出。首先,为了突出显示聚类分区y,plotFateMap可以生成与使用的颜色方案相同的颜色方案(或者,可以使用用户定义的任意方案执行这两个功能):
set.seed(111111) fcol <- sample(rainbow(max(y)))
现在,procsom的不同输出数据可以绘制。
从SOM派生的所有模块的平均z分数:
plotheatmap(ps$nodes.z, xpart=y[n], xcol=fcol, ypart=unique(ps$nodes), xgrid=FALSE, ygrid=TRUE, xlab=FALSE)

SOM模块中每个基因的z-score概况:
plotheatmap(ps$all.z, xpart=y[n], xcol=fcol, ypart=ps$nodes, xgrid=FALSE, ygrid=TRUE, xlab=FALSE)

SOM模块排序的每个基因的标准化表达谱:
plotheatmap(ps$all.e, xpart=y[n], xcol=fcol, ypart=ps$nodes, xgrid=FALSE, ygrid=TRUE, xlab=FALSE)

每个基因的二值化表达谱(z-score <-1,-1<z-score <1,z-score="" style="font-size: inherit; color: inherit; line-height: inherit;">1):
plotheatmap(ps$all.b, xpart=y[n], xcol=fcol, ypart=ps$nodes, xgrid=FALSE, ygrid=TRUE, xlab=FALSE)
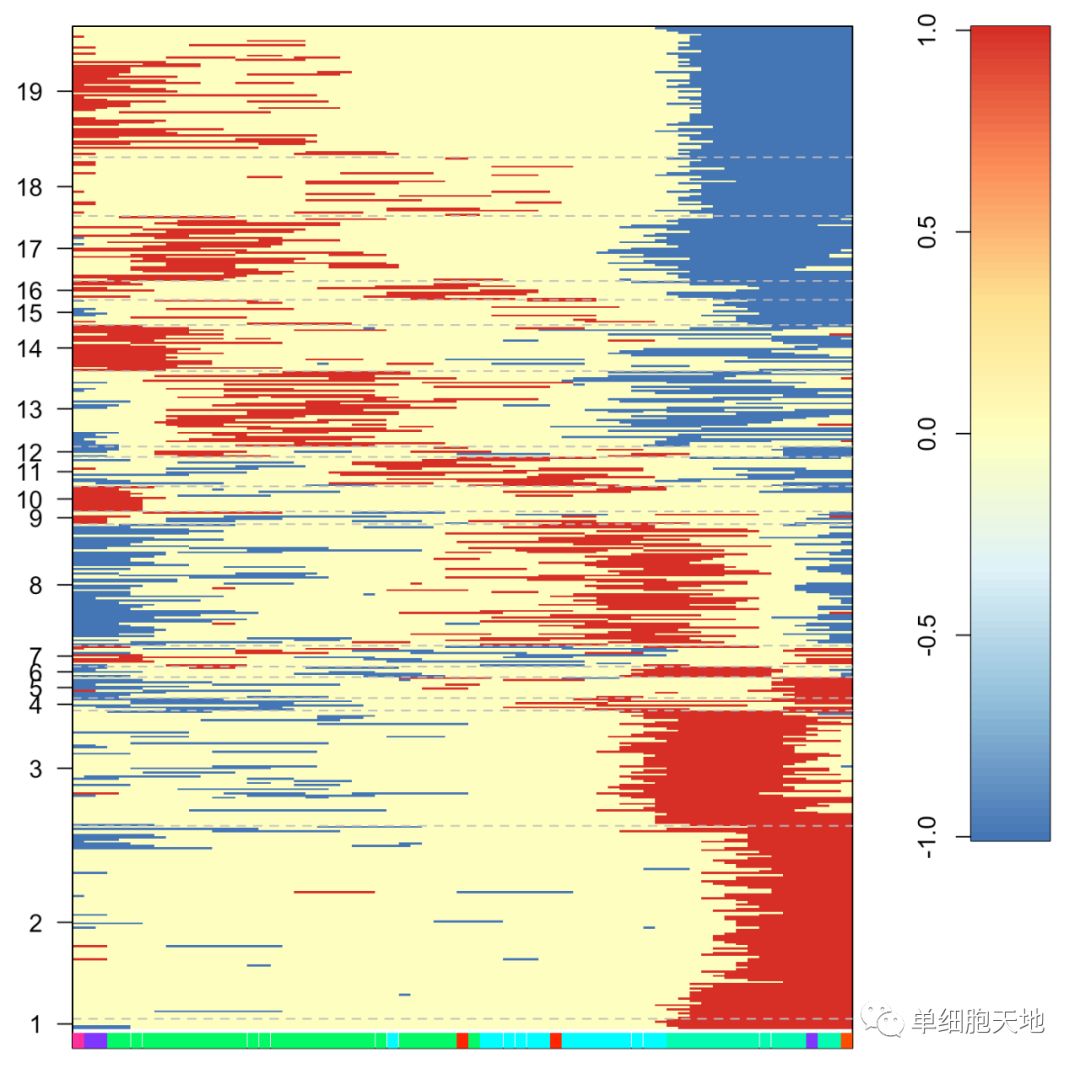
为了检查SOM的各个模块内的基因,可以在给定模块的数量的情况下提取这些基因。模块编号包含在procsom函数的返回参数节点中,并且可以提取,例如,对于模块编号1:
g <- names(ps$nodes)[ps$nodes == 1]
该组的平均伪时间表达谱可以通过函数plotexpression绘制:
plotexpression(fs, y, g, n, col=fcol, name="Node 1", cluster=FALSE, alpha=.5, types=NULL)

以同样的方式可以绘制单个基因的表达谱,例如:
plotexpression(fs, y, "Clca4__chr3", n, col=fcol, cluster=FALSE, alpha=.5, types=NULL)

也可以使用types参数将数据点突出显示为特定符号,例如反映批次:
plotexpression(fs, y, g, n, col=fcol, name="Node 1", cluster=FALSE, types=sub("\_\d+","",n))

差异基因表达分析
FateID还提供了差异基因表达分析的功能。该功能实现了类似于DESeq (Anders和Huber 2010)的方法,通过该方法将负二项分布拟合到两个群体中的转录物计数,例如表示两种不同的细胞类型。默认情况下,该函数应在标准化数据上执行,并根据对数转换的转录计数方差的多项式拟合估计色散参数,作为对数转换平均表达式的函数。这是在输入数据上完成的,要么在汇集来自两个群体的细胞之后,要么分别在每个群体上汇集细胞。或者,还可以提供方差 – 平均依赖性的函数作为输入参数,例如在RaceID2中计算的依赖性(Grun et al.2016)异常值识别。作为另一种选择,该功能还可以执行标准DESeq2 (Love,Huber和Anders 2014)差异表达分析。
为了运行分析,表达数据与cell IDs向量对应于此数据集的列名子集要一起输入。
例如,该功能可用于鉴定原始簇3,4,5(祖细胞区室)中细胞之间差异表达的基因,这些细胞偏向于肠细胞(簇6)或杯状细胞(簇13),因为命运概率> 0.5:
thr <- .5 a <- "t13" b <- "t6" cl <- c(3,4,5) A <- rownames(fb$probs)[fb$probs[,a] > thr] A <- A[y[A] %in% cl] B <- rownames(fb$probs)[fb$probs[,b] > thr] B <- B[y[B] %in% cl] de <- diffexpnb(v,A=A,B=B,DESeq=FALSE,norm=FALSE,vfit=NULL,locreg=FALSE)
在这里,我们使用没有进行特征选择的完整表达数据作为输入。不执行标准化,因为v已经按大小标准化。利用所选择的输入参数,diffexpnb将基于RaceID2(Grun等人2016)中导出的方差 – 均值依赖性推断两个群体的负二项转录物水平分布的分散参数。或者,如果locreg等于TRUE,则可以使用局部回归。另一种选择是运行DESeq2 (Love,Huber和Anders 2014)分析,如果DESeq是TRUE的话。该函数返回包含三个部分的列表。差异基因表达分析的结果存储在res组件中,res组件是显示两组平均表达的数据框,两组之间的倍数变化和log2倍数变化,差异表达式的p值(pval)和Benjamini-Hochberg纠正了错误发现率(padj)。
结果可以通过以下函数绘制:
plotdiffgenesnb(de,mthr=-4,lthr=0,Aname=a,Bname=b,padj=FALSE)

检查基因表达的命运偏见
为了能够进一步检查命运特异性标记基因表达,FateID提供了在散点图中绘制两个基因彼此表达水平的功能。例如,这些基因可以代表两种不同谱系的标记,并且该功能揭示这些基因的表达域是否落入不同的簇中,或者它们是否与命运偏向特定谱系相关。为了可视化基因表达域和细胞簇起源的相关性,gene2gene可以用基因表达数据框和聚类分区作为输入来调用该函数:
gene2gene(intestine$v,intestine$y,"Muc2__chr7","Apoa1__chr9")
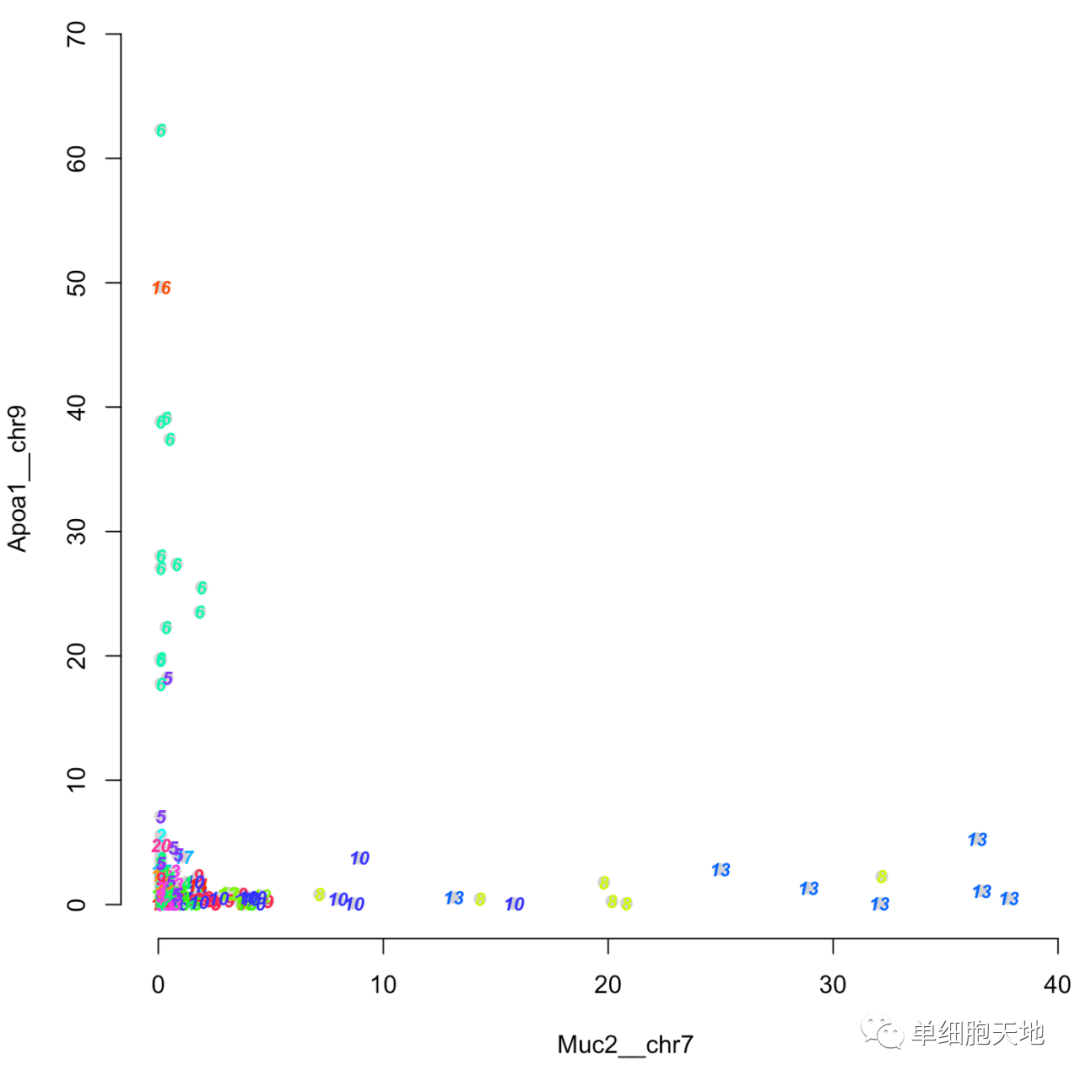
如果向特定的目标群的命运偏移,例如群集6,fateBias的输出fb和fb$probs的列名对应于目标群集,即t6,需要作为附加输入参数:
gene2gene(intestine$v, intestine$y, "Muc2__chr7", "Apoa1__chr9", fb=fb, tn="t6", plotnum=FALSE)

为了识别在分化的所有阶段参与早期启动和命运决定的基因,提取具有高重要性值的所有基因(参见randomForest包中的randomForest函数的详细信息)以分类到至少在一次随机森林迭代中给定的目标聚类中:
k <- impGenes(fb,"t6")

此函数需要函数fateBias的输出对象和目标集群的数量,并与t连接作为输入。其他可选参数指定重要性值的截止值(参见手册页)。它返回含有两个对象的列表,具有所有基因的平均重要性值的一个数据集,这里所述基因至少在迭代中作为行或作为列通过阈值,以及具有重要性值的标准偏差的相应数据集。
该函数还绘制了重要值的热图,其中基因进行了层次聚类。
References
Anders, S., and W. Huber. 2010. “Differential expression analysis for sequence count data.” Genome Biol. 11 (10): R106.
Angerer, P., L. Haghverdi, M. Buttner, F. J. Theis, C. Marr, and F. Buettner. 2016. “destiny: diffusion maps for large-scale single-cell data in R.” Bioinformatics 32 (8): 1241–3.
Breiman, L. 2001. “Random Forests.” Mach. Learn. 45 (1): 5–32.
Grun, D., M. J. Muraro, J. C. Boisset, K. Wiebrands, A. Lyubimova, G. Dharmadhikari, M. van den Born, et al. 2016. “De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data.” Cell Stem Cell 19 (2): 266–77.
Herman, J. S., Sagar, and D. Grün. 2018. “FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data.” Nat. Methods 15 (5): 379–86.
Love, M. I., W. Huber, and S. Anders. 2014. “Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2.” Genome Biol.15 (12): 550.
Maaten, L. van der, and G. Hinton. 2008. “Visualizing Data using t-SNE.” J. Mach. Learn. 9: 2570–2605.
Qiu, X., A. Hill, J. Packer, D. Lin, Y. A. Ma, and C. Trapnell. 2017. “Single-cell mRNA quantification and differential analysis with Census.” Nat. Methods 14 (3): 309–15.


