单细胞分析实录(9): 展示marker基因的4种图形(二)
在上一篇中,我已经讲解了展示marker基因的前两种图形,分别是tsne/umap图、热图,感兴趣的读者可以回顾一下。这一节我们继续学习堆叠小提琴图和气泡图。
3. 堆叠小提琴图展示marker基因
相比于其他可视化形式,小提琴图可以更直观地展示某一类亚群的某一个基因的表达分布情况。我的marker基因一共选了12个,下面来画图:
Seurat内置的VlnPlot函数可以直接画,
library(xlsx)
markerdf2=read.xlsx("ref_marker2.xlsx",sheetIndex = 1)
markerdf2$gene=as.character(markerdf2$gene)
mye.seu=readRDS("mye.seu.rds")
mye.seu$celltype=factor(mye.seu$celltype,levels = sort(unique(mye.seu$celltype)))
Idents(mye.seu)="celltype"
VlnPlot(mye.seu, features = markerdf2$gene, pt.size = 0, ncol = 1)+
scale_x_discrete("")+
theme(
axis.text.x.bottom = element_blank()
)
ggsave("vln1.pdf",width = 20,height = 80,units = "cm")
其中pt.size参数表示点的大小,一个点就是一个细胞,一般可以直接设置为0,即不显示点,只画小提琴,看上去更加清楚。尽管此处我对标度和主题进行了调整,但我发现这只对单个feature有用,多个feature时就不起作用了,后续就用AI来简单编辑一下吧。
需要注意的是,图的颜色是根据亚群的类别来划分的,并不是根据基因来区分。
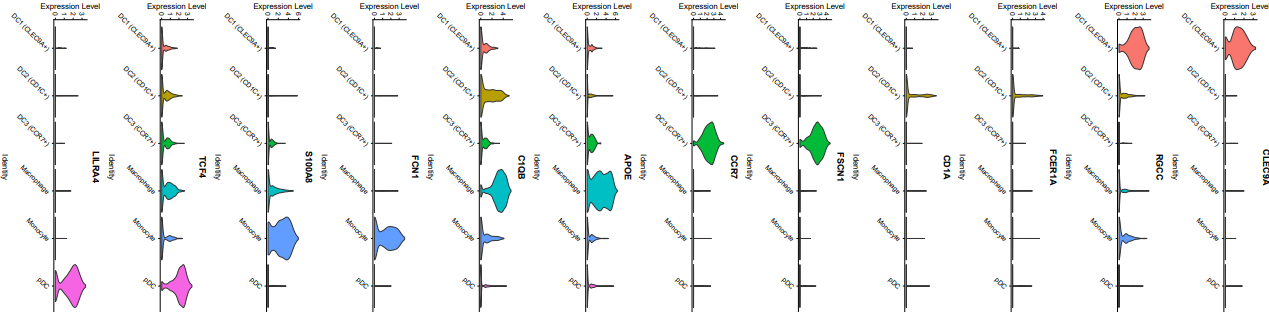
第二种方法,ggplot2代码如下
library(reshape2)
vln.df=as.data.frame(mye.seu[["RNA"]]@data[markerdf2$gene,])
vln.df$gene=rownames(vln.df)
vln.df=melt(vln.df,id="gene")
colnames(vln.df)[c(2,3)]=c("CB","exp")
#数据格式如下
# > head(vln.df)
# gene CB exp
# 1 CLEC9A N01_AAACGGGCATTTCAGG_1 0.000
# 2 RGCC N01_AAACGGGCATTTCAGG_1 0.000
# 3 FCER1A N01_AAACGGGCATTTCAGG_1 0.000
# 4 CD1A N01_AAACGGGCATTTCAGG_1 0.000
# 5 FSCN1 N01_AAACGGGCATTTCAGG_1 1.104
# 6 CCR7 N01_AAACGGGCATTTCAGG_1 0.000
[email protected][,c("CB","celltype")]
vln.df=inner_join(vln.df,anno,by="CB")
vln.df$gene=factor(vln.df$gene,levels = markerdf2$gene) #为了控制画图的基因顺序
vln.df%>%ggplot(aes(celltype,exp))+geom_violin(aes(fill=gene),scale = "width")+
facet_grid(vln.df$gene~.,scales = "free_y")+
scale_fill_brewer(palette = "Set3",direction = 1)+
scale_x_discrete("")+scale_y_continuous("")+
theme_bw()+
theme(
axis.text.x.bottom = element_text(angle = 45,hjust = 1,vjust = 1),
panel.grid.major = element_blank(),panel.grid.minor = element_blank(),
legend.position = "none"
)
ggsave("vln2.pdf",width = 11,height = 22,units = "cm")
geom_violin()函数的scale参数为”width”时,所有小提琴有相同的宽度,默认是”area”,有相同的面积;facet_grid()用来分面,文中用的是多行一列,scales = “free_y”表示不同行之间可以有不同范围的y值;scale_fill_brewer()使用ColorBrewer调色板。
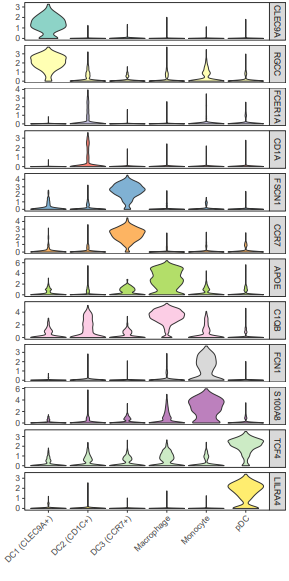
这个图的颜色根据基因来区分,有时可能还会看到小提琴图的颜色是用亚群某个基因的表达均值来映射的,比如
vln.df$celltype_gene=paste(vln.df$celltype,vln.df$gene,sep = "_")
stat.df=as.data.frame(vln.df%>%group_by(celltype,gene)%>%summarize(mean=mean(exp)))
stat.df$celltype_gene=paste(stat.df$celltype,stat.df$gene,sep = "_")
stat.df=stat.df[,c("mean","celltype_gene")]
vln.df=inner_join(vln.df,stat.df,by="celltype_gene")
vln.df$mean=ifelse(vln.df$mean > 3, 3, vln.df$mean)
#这里的阈值3要提前综合所有基因看一下
vln.df%>%ggplot(aes(celltype,exp))+geom_violin(aes(fill=mean),scale = "width")+
facet_grid(vln.df$gene~.,scales = "free_y")+
scale_fill_gradient(limits=c(0,3),low = "lightgrey",high = "yellow")+
scale_x_discrete("")+scale_y_continuous("",expand = c(0.02,0))+
theme_bw()+
theme(
panel.grid.major = element_blank(),panel.grid.minor = element_blank(),
axis.text.x.bottom = element_text(angle = 45,hjust = 1,vjust = 1)
)
ggsave("vln3.pdf",width = 11,height = 22,units = "cm")
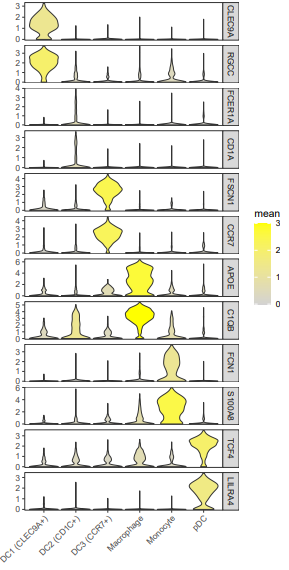
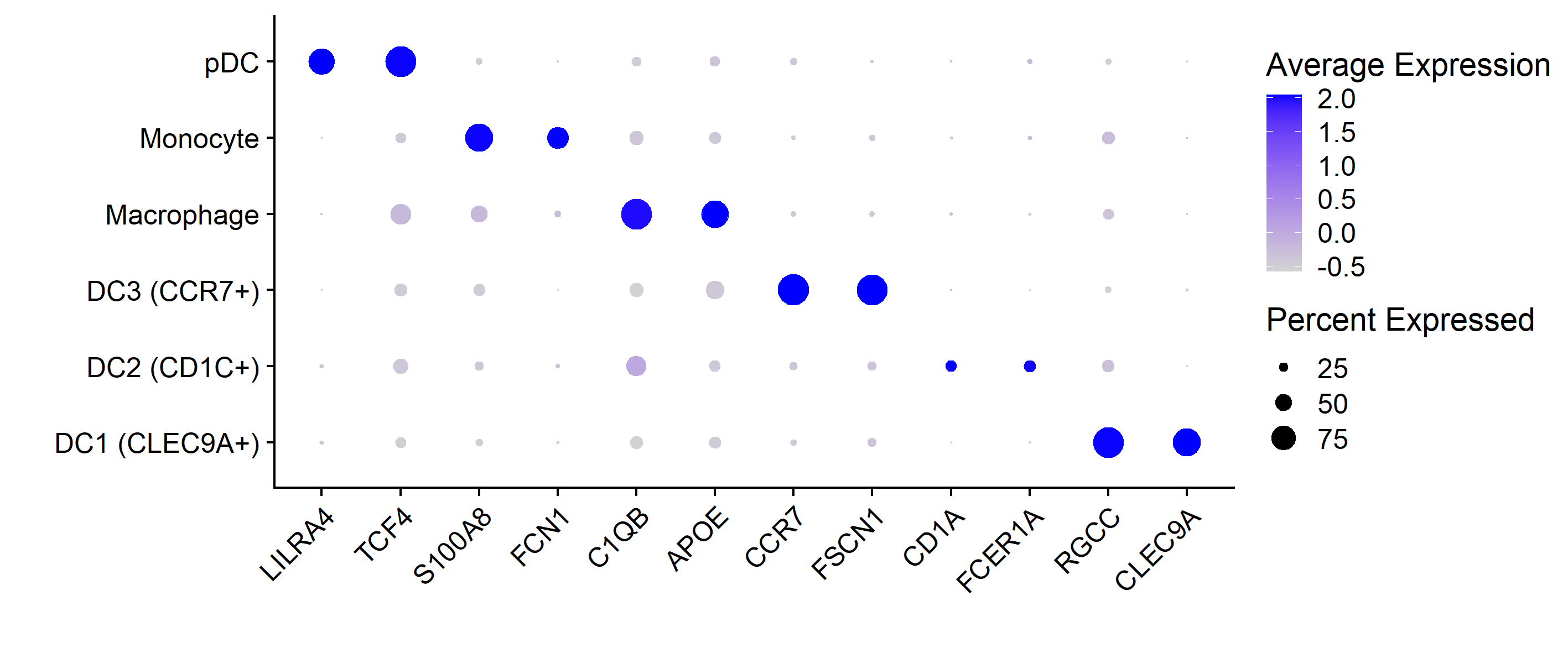
4. 气泡图展示marker基因
Seurat的画法是这样的,
DotPlot(mye.seu, features = markerdf2$gene)+RotatedAxis()+
scale_x_discrete("")+scale_y_discrete("")
#其余的微调同ggplot2
第二种方法,ggplot2代码如下
bubble.df=as.matrix(mye.seu[["RNA"]]@data[markerdf2$gene,])
bubble.df=t(bubble.df)
bubble.df=as.data.frame(scale(bubble.df))
bubble.df$CB=rownames(bubble.df)
bubble.df=merge(bubble.df,[email protected][,c("CB","celltype")],by = "CB")
bubble.df$CB=NULL
celltype_v=c()
gene_v=c()
mean_v=c()
ratio_v=c()
for (i in unique(bubble.df$celltype)) {
bubble.df_small=bubble.df%>%filter(celltype==i)
for (j in markerdf2$gene) {
exp_mean=mean(bubble.df_small[,j])
exp_ratio=sum(bubble.df_small[,j] > min(bubble.df_small[,j])) / length(bubble.df_small[,j])
celltype_v=append(celltype_v,i)
gene_v=append(gene_v,j)
mean_v=append(mean_v,exp_mean)
ratio_v=append(ratio_v,exp_ratio)
}
}
plotdf=data.frame(
celltype=celltype_v,
gene=gene_v,
exp=mean_v,
ratio=ratio_v
)
plotdf$celltype=factor(plotdf$celltype,levels = sort(unique(plotdf$celltype)))
plotdf$gene=factor(plotdf$gene,levels = rev(as.character(markerdf2$gene)))
plotdf$exp=ifelse(plotdf$exp>3,3,plotdf$exp)
plotdf%>%ggplot(aes(x=celltype,y=gene,size=ratio,color=exp))+geom_point()+
scale_x_discrete("")+scale_y_discrete("")+
scale_color_gradientn(colours = rev(c("#FFD92F","#FEE391",brewer.pal(11, "Spectral")[7:11])))+
scale_size_continuous(limits = c(0,1))+theme_bw()+
theme(
axis.text.x.bottom = element_text(hjust = 1, vjust = 1, angle = 45)
)
ggsave(filename = "bubble2.pdf",width = 9,height = 12,units = c("cm"))
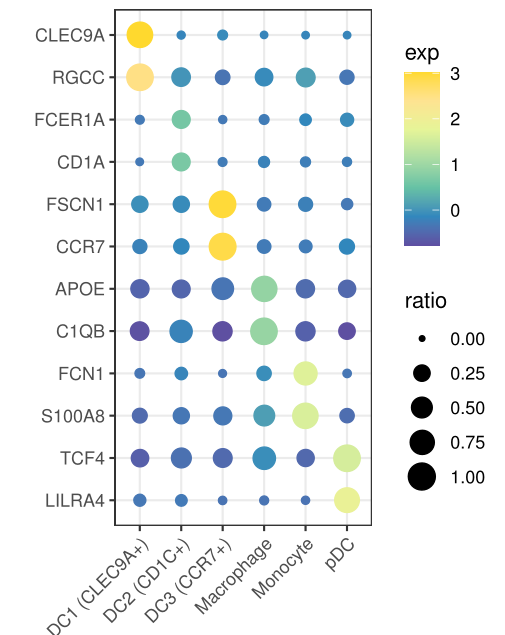
这两种方法具体函数定义略有差异,所以气泡图看上去不太一样
到这里,marker基因的可视化就结束了,基本就是这些。如果你觉得上述内容对你有用,欢迎转发,点赞!有任何疑问可以在公众号后台提出,我都会回复的。
因水平有限,有错误的地方,欢迎批评指正!


